Cancer Adaptive Therapy Models
#CATMO2020
Cancer Adaptive Therapy Models
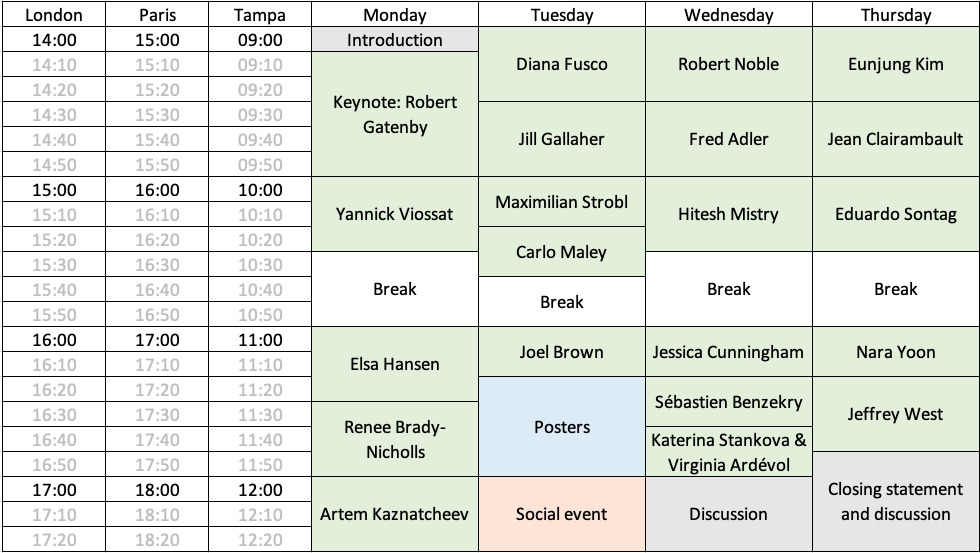
We are organizing an online workshop on Models of Adaptive Therapy. It will be held from Dec. 7th to 10th, 2020, just three hours a day (9-12 am, US Eastern time, 3-6 pm European continental time), in order to facilitate attendance by both Europeans and American researchers.
Adaptive Cancer Therapy
Treatment resistance in cancer therapy remains an overarching challenge across all types of cancer and modes of treatment including targeted therapy, chemotherapy, and immunotherapy. Despite the ubiquity of the evolution of resistance, the “more is better'' paradigm prevails in standard of care approaches. Over the past decade, a small group of oncologists in collaboration with evolutionary and experimental biologists have proposed an adaptive approach to cancer treatment.
Mathematical Modeling
We aim at gathering the main groups that have been recently trying to model adaptive therapy using mathematics, or whose work could help to do so, in order to build a community, understand the state of the art, exchange on the directions the field should take, and foster collaborations. On top of regular talks and a poster session, time will be allowed for discussions. Both proponents of and opponents to adaptive therapy are welcome. There are no participation fees (at least for the speakers, and the first participants to register).
In order to help participants to better understand the state of the art, speakers are encouraged to mention not only their work but also related studies by other researchers. Speakers are also encouraged to discuss what could be done next, which kind of collaborations could be helpful, which assumptions they would like to relax, which experiments should be run, or anything that may feed discussions on limitations of our current understanding and future directions for the field.
Logistics
While COVID-19 continues spreading across the world, we are keeping the health and safety of attendees as our top priority. To reduce or slow the spread of infection, this meeting will be held virtually.
To facilitate this virtual setting, we have selected a third-party virtual meeting platform (Sococo) that seamlessly supports live video presentations of sessions, poster sessions, networking and socializing. The site also will allow small groups of participants to interact in real-time directly with each other. To learn how to use Sococo, watch this short training video.
We invite researchers in all career stages to apply to give a talk or poster presentation.
Registration
The deadline to register to attend CATMo has now passed.
Poster submission deadline
Poster presenters should submit their poster (PDF) by email to one of the conference organizers by Friday, the 4th of December.